- Dna to rna conversion python
- Saved searches
- Use saved searches to filter your results more quickly
- CyrusK/convert_dna_to_rna_or_protein.py
- Name already in use
- Sign In Required
- Launching GitHub Desktop
- Launching GitHub Desktop
- Launching Xcode
- Launching Visual Studio Code
- Latest commit
- Git stats
- Files
- README.md
- About
- Dna to rna conversion python
- Intelligent Recommendation
- Use LIMMA, GLIMMA and EDGER, RNA-SEQ data analysis is easy
- Reverse complement of DNA strand using Python
- An overview of DNA and RNA as used in Molecular Biology
- Reverse Complement of a DNA or RNA
- How to identify if the sequences of DNA and RNA
- Method 1: Verify if a sequence is DNA and RNA
Dna to rna conversion python
Write a Python script that translates two genes in an RNA sequence into their protein sequence and prints them. Each gene begins with an AUG from the left and ends in UAG and has a length that is a multiple of three. However, the RNA sequence length may not be a multiple of three and there may be more than one «UAG» or «AUG» in the sequence.
For example if the input is
with open ("p:/dna.txt", "r") as myfile: data=myfile.readlines() map = DNA=data[1] flag = 1 while flag: start = DNA.find('AUG') if start == -1: flag = 0 else: done = 0 while done!= 0: i = start codon = DNA[1:i+3] if codon == "UAG": stop = i protein = translate(DNA(start)) DNA = DNA[stop:] done = 1 print(protein) then the output should be
I have this so far. http://dpaste.org/v2e9/ can anyone help out?
ADD COMMENT • link updated 9.9 years ago by viv_bio ▴ 50 • written 12.8 years ago by Studentguy ▴ 70
@Simon: while I sometimes feel irked when I see a question that seems to be taken right out of a homework I think in the end is not our job to police this. Plus we may be wrong in our assumptions. So I would leave this up to everyone’s individual opinion on whether they would want to answer it or not. A great solution to an answer lives on and will continue to provide value beyond the original poster’s needs.
I do agree, however there is for that question a partial solution if you follow the link to the OP’s ‘dpaste’ page. Here is a thought. With enough googling, the StudentGuy will come up with an already made up solution anyways, most probably using Biopython, which he will likely not understand and which will be too much high order (using ready made package) to have much teaching value. At least here the OP did a part of the work and is ready to interact with people who likely will teach him something. A better developped question and including the code right here might have been better. Cheers
I’d be interested in other moderators opinion of homework questions? I think proof of a reasonable stab at a solution would be a good thing, rather than ‘do my homework for me’ style questions.
just need to find an otherwise permissive license that prohibits copy-paste use into a homework solution
@brentp: The people who are copy-pasting homework probably aren’t reading enough to look at the licenses anyways.
some consolation: I appreciates the frankness to write it as HW!!
I am not really looking for handouts but I am new to programming and am occasionally very confused with python. Most uni’s now require all bio majors to be thrown into the pit with a BNFO class since even with limited knowledge in the subject it is a valuable skill to have. that being said all of our teachers here are pretty seasoned vets in several languages and sometimes its hard to connect to «my first programming class»
i posted more below in the answers section 😀
homework? well, i’ll answer anyway using biopython.
from Bio.Seq import Seq from Bio.Alphabet import generic_rna # add your own logic here to parse the rna sequence from the file. # split on start codon. drop the part preceding the 1st start codon, # then for each chunk, translate to the stop codon. then join and print. print " ".join((str(Seq("AUG" + rest, generic_rna).translate(to_stop=True)) for rest in "ACAUGCUAGAAUAGCCGCAUGUACUAGUUAA".split("AUG")[1:])) I’m not a python guy but the following script does the job with dna.txt=
>Human ACAUGCUAGAAUAGCCGCAUGUACUAGUUAA src: you just need to check if there is at least 3 bases available after the current position:
with open ("dna.txt", "r") as myfile: data=myfile.readlines() map = DNA=data[1].strip() start = DNA.find('AUG') if start!= -1: while start+2 < len(DNA): codon = DNA[start:start+3] if codon == "UAG": break; print(map[codon]) start+=3 ADD COMMENT • link updated 3.9 years ago by Ram 39k • written 12.8 years ago by Pierre Lindenbaum 155k
hi i ran your code but there seem to be a problem with your code. When i read a data file n characters are also read. And the start variable seem to always get the value -1 even when the sub-string is present in DNA.
when I run your code I get this error:
start, end = next_transcript(mRNA, cur_pos) TypeError: 'NoneType' object is not iterable I also noticed that cur_pos isn't defined until later line 43. Could this be the problem? I am also not sure why in DNA=data[1].strip() you call for the second item 1 in the string? How is your dna.txt formated? This clarification would be much appreciated. Thanks
Thank you all for the fast responses, and to those who help i greatly appreciate it, i ended up with an alternate solution.
I am not really looking for handouts but I am new to programming and am occasionally very confused with python. Most uni's now require all bio majors to be thrown into the pit with a BNFO class since even with limited knowledge in the subject it is a valuable skill to have. that being said all of our teachers here are pretty seasoned vets in several languages and sometimes its hard to connect to "my first programming class"
heres the code i ended up using, it did away with that loop structure i couldnt get and kinda cheats by using find/rfind two narrow down two sequences, it wont work for more than two sequences but it does the job none the less
Nice work with this. Your logic is sound and the major remaining issue to tackle is repetitiveness in the code. Whenever you repeat code with a few changes, you should focus on making that code into a function. Here's a next version of your code generalized with functions and a while loop. It's a combination of your first and second attempts. Notice how the functions allow you to avoid re-writing the same code multiple times: http://gist.github.com/626765
from Bio import SeqIO , Seq from Bio.SeqRecord import SeqRecord def make_trans_record(record): "Returns a new seqrecord with translated sequences" return SeqRecord(seq = record.seq[350:-103].translate(),\ description = "") Input = raw_input("Enter File location of the nucleotide sequence :") Output = raw_input("Output file location and name :") records = map(make_trans_record,SeqIO.parse(Input,"fasta")) SeqIO.write(records,Output,"fasta") Saved searches
Use saved searches to filter your results more quickly
You signed in with another tab or window. Reload to refresh your session. You signed out in another tab or window. Reload to refresh your session. You switched accounts on another tab or window. Reload to refresh your session.
Convert any DNA string into an RNA translation or a theoretically valid Protein sequence using Python that takes an operation and filename from the command line.
CyrusK/convert_dna_to_rna_or_protein.py
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
Name already in use
A tag already exists with the provided branch name. Many Git commands accept both tag and branch names, so creating this branch may cause unexpected behavior. Are you sure you want to create this branch?
Sign In Required
Please sign in to use Codespaces.
Launching GitHub Desktop
If nothing happens, download GitHub Desktop and try again.
Launching GitHub Desktop
If nothing happens, download GitHub Desktop and try again.
Launching Xcode
If nothing happens, download Xcode and try again.
Launching Visual Studio Code
Your codespace will open once ready.
There was a problem preparing your codespace, please try again.
Latest commit
Git stats
Files
Failed to load latest commit information.
README.md
Convert DNA to RNA or Protein
Here is some Python script I put together (DNA translation file above) that takes any DNA string (gDNA, cDNA, etc..) and optionally prints its RNA translation or all of the possible valid protein(s) sequences with start and stop codons included. The script will conveniently take an operation and filename of your choice from the command line and print the output.
$dna.txt GCGCTTGGTTTAATGACGGCTTGTTTCTTTTCTGTGGCTGCGTGAAAGCCTTGAGGGGCTCCGGGAGGGCCCTTT GTGCGGGGGGAGCGGCTCGGGGGGTGCGTGCGTGTGTGTGTGCGTGGGGAGCG
$python conversion.py --rna dna.txt GCGCUUGGUUUAAUGACGGCUUGUUUCUUUUCUGUGGCUGCGUGAAAGCCUUGAGGGGCUCCGGGAGGGCCCUUU GUGCGGGGGGAGCGGCUCGGGGGGUGCGUGCGUGUGUGUGUGCGUGGGGAGCG
$python conversion.py --genes dna.txt
1 valid protein(s)can be produced from this sequence: DNA: ATGACGGCTTGTTTCTTTTCTGTGGCTGCGTGA RNA: AUGACGGCUUGUUUCUUUUCUGUGGCUGCGUGA AA sequence: MTACFFSVAA
The code in this repository is open source. It may be useful for anyone in biotech R&D working on big data genomic analysis.
About
Convert any DNA string into an RNA translation or a theoretically valid Protein sequence using Python that takes an operation and filename from the command line.
Dna to rna conversion python
The idea for the conclusion is to replace the T in the DNA sequence with U, just import re directly, and then use sub.
import re rna_nucleotide = '' with open(r'D:\Rosalind\haha.txt','w+') as f1: with open(r'D:\Rosalind\rosalind_rna.txt','r') as f: for nucleotide in f: rna_nucleotide += re.sub('T','U',nucleotide) print (rna_nucleotide,file = f1)
Or in the interactive interface, import re, then re.sub('T','U',seq), is more convenient, but the premise is that the sequence is short, and the memory is not enough if the sequence is long.
Intelligent Recommendation

Use LIMMA, GLIMMA and EDGER, RNA-SEQ data analysis is easy
Contents 1 summary 2 background introduction 3 initial configuration 4 data integration 4.1 Reading count data 4.2 Organize sample information 4.3 Organizational Gene Notes 5 data pretreatment 5.1 Ori.
Reverse complement of DNA strand using Python
In this article, we will cover, how to Reverse the complement of DNA or RNA sequences in Python.
DNA strand: ATGCCGAGCA Complementary Strand: TACGGCTCGT Reverse-Complementary strand: ACGAGCCGTA
An overview of DNA and RNA as used in Molecular Biology
The genetic material of living organisms is made up of Deoxyribonucleic acid(DNA) or Ribonucleic acid (RNA). The primary structure of DNA and RNA is made up of a sequence of nucleotide bases. The structure of DNA can be a double-stranded or single-stranded sequence of nucleotides(bases). For double-stranded nucleic acids, the nucleotide bases pair in a given rule which is unique to DNA and RNA. For DNA, there exist four types of bases namely; Adenine(A), Thymine(T), Guanine(G), and Cytosine(C). Therefore, DNA can be identified as containing ATGC bases. The pairing of bases in DNA is that Adenine pairs with Thymine(with a double bond) while Guanine Pairs with Cytosine (with a triple bond). i.e A=T and G≡C as shown below.
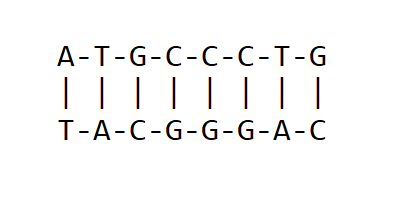
DNA base pairing. The upper strand is complementary to the downer strand and vice versa
For RNA, all instances of Thymine are replaced by Uracil. This means that for double-stranded RNA, Adenine pairs with Uracil while Guanine pairs with Cytosine A=U and G≡C as shown below:
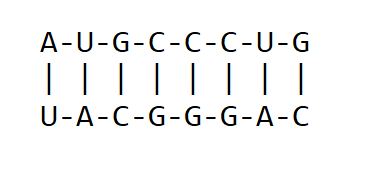
RNA base pairing. Each strand is a complementary sequence to one another
Reverse Complement of a DNA or RNA
A Reverse Complement converts RNA or DNA sequence into its reverse, complement counterpart. One of the major questions in Molecular Biology to solve using computational approaches is to find the reverse complement of a sequence. This is always done so to work with the reversed-complement of a given sequence if it contains an open reading frame(a region that encodes for a protein sequence during the transcription process) on the reverse strand. One could be interested to verify that the sequence is a DNA or RNA before finding its reverse complement
How to identify if the sequences of DNA and RNA
One of the major tasks in Bioinformatics in computational molecular biology and bioinformatics is to verify if the sequence is DNA or RNA. To do this we can use the set method to verify a sequence.
Method 1: Verify if a sequence is DNA and RNA
In the set method, we convert the input sequence into a set. We combine the set obtained with a reference DNA set(ATGC) or RNA set(AUGC) using the union function of the set. This is done so that the input sequence is rendered valid even if it does not contain all four types of nucleotide bases. For instance, TTTTTTTAAA is a valid DNA even though it contains only two types of bases. Also, UUUUUUUUGGG is a valid RNA.